- 1.How to use FluPhenotype?
-
Firstly, input the genomic sequences or proteome of the virus. Attention, one genome or proteome is allowed in each input box. More genome/proteome can be added by click the button “Add another virus genome”.

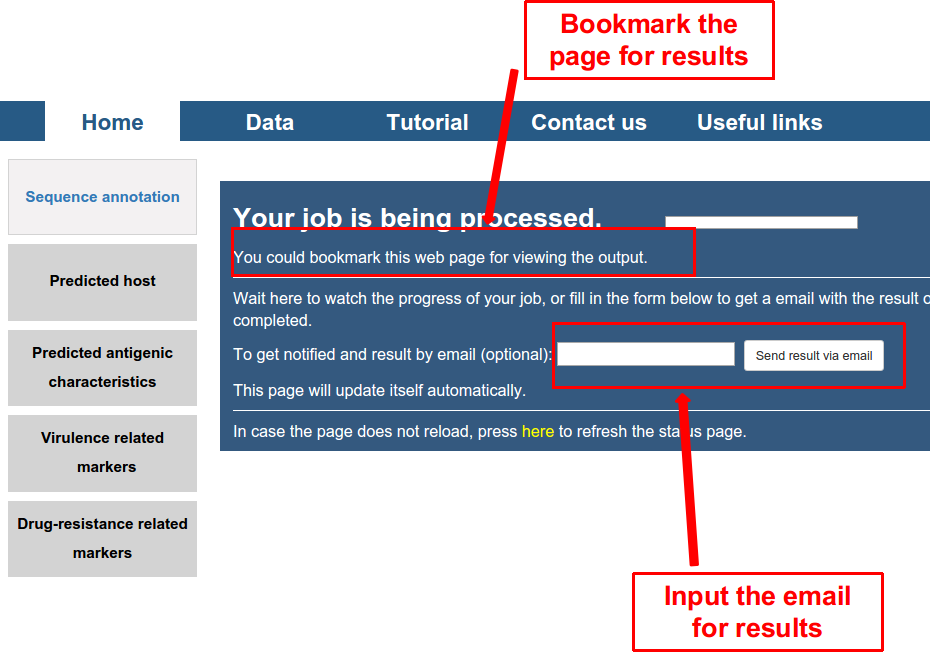
Secondly, submit the task by clicking on the button of “GO”, and a waiting page appeared as follows. User can wait here. It would last for several seconds to several minutes depending on the number of viral genomes inputted. The user can also bookmark the page or input the email (optional), and check the results later.
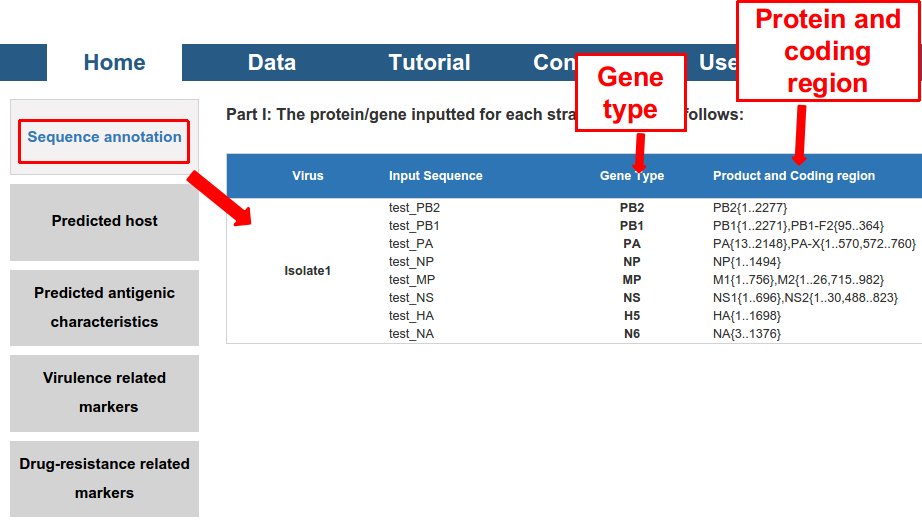
Thirdly, five kinds of output were displayed. All of them can be exported as a PDF file.
1) the “Sequence annotation”, including the gene/protein type and coding region of the sequence (for DNA).
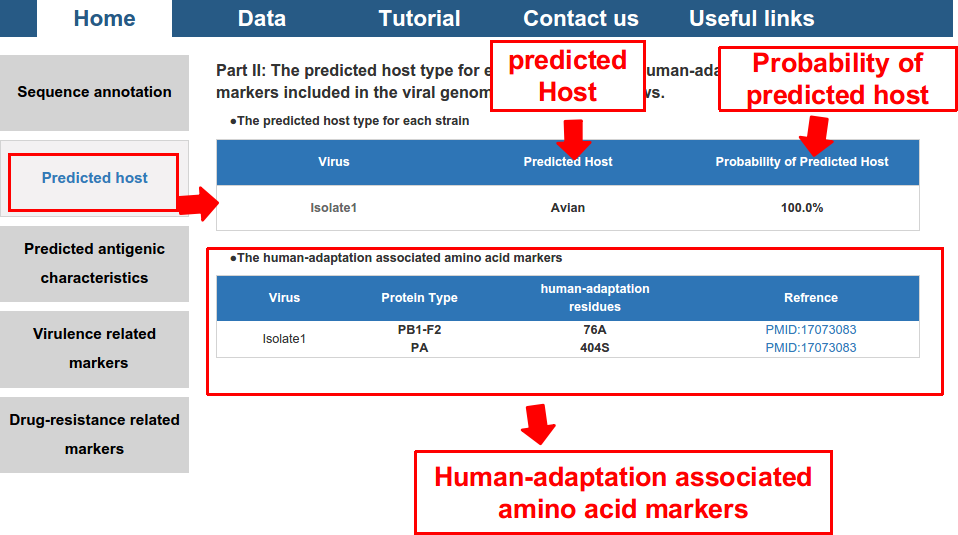
2) predicted potential host and its probability, and also the human-adaptation related amino acid markers included in the proteome of the virus.
3) antigenic and genetic relationship with the vaccine strains of the HA subtype.

4) mammalian virulence related amino acid markers included in the proteome of the virus.

5) drug-resistance (including Amantadine, Oseltamivir, Zanamivir, and baloxavir marboxil) related amino acid markers included in the proteome of the virus.

- 2.Browser compatibility of FluPhenotype
-
OS Version Chrome Firefox Microsoft Edge Safari Linux Ubuntu 69.0.3497.81 63.0.3 n/a n/a MacOS Mojave 70.0.3538.110 63.0.3 n/a 12.0.1 Windows 10 71.0.3578.80 63.0.3 42.17134.1.0 n/a
- 3.How to infer the antigenic relationship between influenza viruses based on HA protein sequences?
- PREDAV-like methods were taken to infer the antigenic relationship between influenza viruses based on HA protein sequences. PREDAV is a naive bayes classifier for predicting antigenic relationship between influenza viruses of H3N2 based on HA protein sequences, which was developed in a previous study (Du et al, Nature Communications, 2012). Here, PREDAV was used to predict the antigenic relationship between influenza H3N2 viruses; PREDAV-H1, which was developed in Liu’s study(Liu et al., Scientific Reports, 2015), was used to predict that of influenza H1N1 viruses; PREDAV-H5, which was developed in Peng’s study (Peng et al., Vaccine, 2014), was used to predict that of influenza H5N1 viruses; PREDAV-FluA, which was developed in Peng’s study (Peng et al., Scientific Reports, 2017), was used to predict that of other types of influenza A viruses.
- 4.What does the odds ratio mean?
- The odds ratio measures the antigenic similarity between viruses. If the odds ratio is smaller than 1, the antigenic relationship of the two viruses is regarded as antigenically similar, otherwise as antigenically distinct. The greater the odds ratio is, the more likely it is that the two viruses are antigenically distinct. Odds ratio of “min” refer to that they were most antigenic similar, while that of “max” refer to that they were most antigenic distinct.
- 5.How to predict the host of influenza viruses on FluPhenotype?
- According our previous studies (Xu et al., PeerJ, 2017), the blast best hit method performed better than HMM best hit method, or the machine learning methods, in predicting the host (avian, human or swine) of influenza viruses based on sequence features. Therefore, we used the blast best hit method for inferring the host of influenza viruses based on sequences. For each viral isolate, each sequence (either nucleotide or protein sequence) of the virus was used to predict the host of the virus based on the blast best hit method, and a voting method was used to determine the most likely host for the virus.
- 6.How to cite us?
-
If you use this server, please cite us in your paper as the following: Youson Peng & Congyu Lu, FluPhenotype: a one-stop platfrom for influenza virus early warnings. Available at
http://www.computationalbiology.cn:18888/IVEW/home
- 7.Reference
-
1.Xiangjun Du#, Libo Dong#, Yu Lan, Yousong Peng, Aiping Wu, Ye Zhang, Weijuan Huang, Dayan Wang, Min Wang, Yuanji Guo, Yuelong Shu* & Taijiao Jiang* (2012). Mapping of H3N2 influenza antigenic evolution in China reveals a strategy for vaccine strain recommendation. Nature communications|3:709| DOI: 10.1038/ncomms1710
2.Xu Beibei, Tan Zhiying, Li Kenli, Jiang Taijiao*, Peng Yousong* (2017), Predicting the host of influenza viruses based on the word vector. PeerJ 5:e3579; DOI10.7717/peerj.3579
3.Yousong Peng#, Xiaodan Li#, Hongbo Zhou, Aiping Wu, Libo Dong, Ye Zhang, Rongbao Gao, Hong Bo, Lei Yang, Dayan Wang, Xian Lin, Meilin Jin*, Yuelong Shu*, Taijiao Jiang*. Continual Antigenic Diversification in China leads to global antigenic complexity of Avian Influenza H5N1 Viruses. Scientific Reports,2017,7:43566,DOI: 10.1038/srep43566
4.Mi Liu#, Xiang Zhao#, Sha Hua#, Xiangjun Du, Yousong Peng, Xiyan Li, Yu Lan, Dayan Wang, Aiping Wu*,Yuelong Shu*, Taijiao Jiang*. Antigenic Patterns and Evolution of the Human Influenza A (H1N1) Virus. Scientific Reports 2015, 5.
5.Yousong Peng#, Yuanqiang Zou#, Honglei Li, Kenli Li, Taijiao Jiang*. Inferring the antigenic epitopes for highly pathogenic avian influenza H5N1 viruses. Vaccine, 2014, 32(6):671-676