

1. What is the antigenic variant for influenza A(H3N2) virus?
Antigenic variant is defined as the virus which has distinct antigenicity with some other virus. Therefore, if virus A is predicted as antigenic variant of virus B, it could escape the immunity generated against virus B.
2. What is the antigenic correlation network?
It reflects the antigenic relationship between viruses. Only if two viruses are predicted to be antigenic similar, they are connected in the network.
3. What is the antigenic cluster?
It is a group of viruses with similar antigenicity (see Du et al, 2012)
4. How the sequences are selected from the database of Influenza Virus Resource?
Except for those generated in the lab through re-assortment, all the human influenza A(H3N2) viruses with HA protein sequence length greater than 290 were collected. Then, their HA protein sequences were aligned. Only the HA1 protein sequences were kept. Finally, those viruses with gap ratio of HA1 protein sequence greater than 10% were removed.
5. How to update the sequences from Influenza Virus Resource and other sources?
The sequences in the database will be updated automatically on the first day of every month to include the newly submitted sequences in the database of Influenza Virus Resource. Besides, we would also manually add the latest sequences from the database of Global Initiative on Sharing Avian Influenza Data (GISAID, available at http://platform.gisaid.org/epi3/frontend#18fa13) into the database. To protect the rights of sequence submitters in GISAID, the information for the sequences derived from GISAID would not be shown in the database. They would only be used in the functions of “Antigenic world map” and “Antigenic cluster dynamics”.
6. How many antigenic clusters were defined for influenza A(H3N2) virus?
Since the appearance of influenza A(H3N2) virus in 1968, there have been lots of predicted antigenic clusters. Here, we mainly consider the dominant antigenic clusters which constitute more than half of viruses in at least a year. In total, 22 dominant antigenic clusters are predicted ever circulating in the world (see the following figure). They are named after the WHO-recommended vaccine strain in the cluster - two letters refer to the location of isolation and two digits refer to year of isolation. The full name of these vaccine strains are listed in the following table.
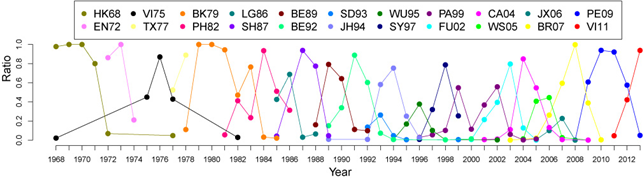
| Abbr | Full name | Abbr | Full name |
|---|---|---|---|
| HK68 | A/HongKong/1/1968 | JH94 | A/Johannesburg/33/1993 |
| EN72 | A/England/42/1972 | WU95 | A/Wuhan/359/1995 |
| VI75 | A/Victoria/3/1975 | SY97 | A/Sydney/5/1997 |
| TX77 | A/Texas/1/1977 | PA99 | A/Panama/2007/1999 |
| BK79 | A/Bangkok/1/1979 | FU02 | A/Fujian/411/2002 |
| PH82 | A/Philippines/2/1982 | CA04 | A/California/7/2004 |
| LG86 | A/Leningrad/360/1986 | WS05 | A/Wisconsin/67/2005 |
| SH87 | A/Shanghai/11/1987 | JX06 | A/Jiangxidonghu/312/2006 |
| BE89 | A/Beijing/353/1989 | BR07 | A/Brisbane/10/2007 |
| BE92 | A/Beijing/32/1992 | PE09 | A/Perth/16/2009 |
| SD93 | A/Shandong/9/1993 | VI11 | A/Victoria/361/2011 |
Except for the dominant antigenic clusters, there are also many minor antigenic clusters, which are named in the form of “Minor-X” (X=1,2,….). For the viruses which are antigenic distinct from all other viruses, they are all named as “Island”.
7. How to recommend influenza vaccine for influenza A(H3N2) virus based on the predicted antigenic clusters?
The spatial-temporal analysis of these predicted antigenic clusters could tell us which antigenic clusters are circulating in the given country/region in the given time period, as shown in the antigenic dynamics plot. If a new antigenic cluster appears in some country with an increasing ratio, it should warn us of its epidemic potential. The detailed method has been described in Du et al, 2012. The following diagram (adapted from Du et al, 2012) shows this process in a vivid way.
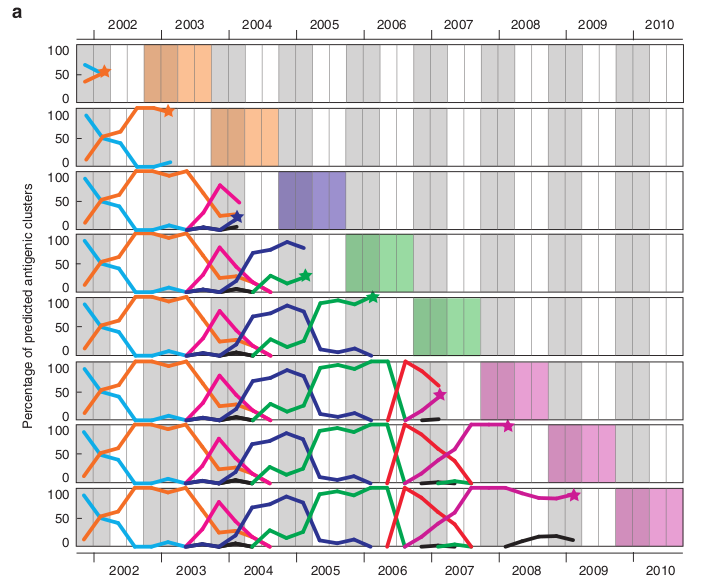
Season by season vaccine strain recommendations for the influenza seasons from 2002-2003 to 2009-2010 based on the H3N2 viruses monitored in China before March 15 of each year from 2002-2009. Dynamic changes in predicted antigenic clusters were monitored by quarter. For clarity, the antigenic clusters prior to 2002 are not shown. Asterisks indicate the dates (March 15 in this case) for vaccine strain recommendation. Gray backgrounds indicate winter seasons in the Northern Hemisphere. The colored backgrounds indicate the season in which the predicted vaccine strains were to be used. Different colored lines represent different clusters.
8. How to predict the antigenic variant based on HA1 protein sequence?
It is predicted with a naive Bayesian classifier developed in Du et al, 2012. The classifier considered 12 structural and physicochemical properties or features that have been reported to or are thought to affect the antigenic properties of influenza HA. These 12 features include five known H3N2 virus HA epitopes, five physicochemical properties of amino acids (hydrophobicity, volume, charge, polarity and accessible surface area), receptor binding and glycosylation. The details of the method could be found in Du et al, 2012.
9. What does the odds ratio mean?
The odds ratio measures the antigenic similarity between viruses. If the odds ratio is smaller than 1, the antigenic relationship of the two viruses is regarded as antigenically similar, otherwise as antigenically distinct. The greater the odds ratio is, the more likely it is that the two viruses are antigenically distinct. Odds ratio equals “min” refer to that they were mostly antigenic similar. Those pairs of viruses not in the table of antigenic and genetic relationship are most antigenic distinct. To reduce space, they are not listed in the table.
10. How to predict the antigenic cluster based on HA1 protein sequence?
It was predicted by the computational method PREDAC, which was developed in Du et al, 2012. It includes three steps, which is shown in the following figure. Firstly, the antigenic relationship (either similar or variant) between viruses is predicted; then, the antigenic similar viruses are used to construct the antigenic correlation network; finally, the antigenic clusters were predicted using the MCl clustering method.
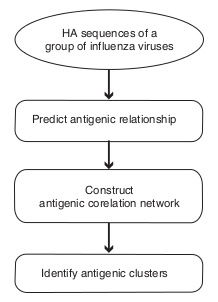
To accurately predict the antigenic clusters for the user input viruses, we firstly determined the most reasonable antigenic clustering for all the human influenza A(H3N2) virus which has HA1 protein sequence available in the Influenza Virus Resources. This antigenic clustering best described the antigenic evolution of H3N2 virus since 1968 (see the antigenic dynamics plot described above). Then, for each dominant antigenic cluster, we randomly selected 100 viruses as the reference virus. They were added to the user input viruses. Because the antigenic clusters they belong to were known in advance, the adjusted rand index was used to determine the best antigenic clustering which matched most with the original antigenic clustering for the reference viruses.
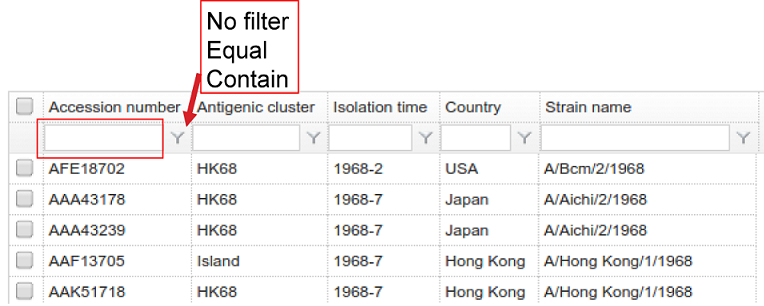
11. How to search the database?
The virus in the database could be searched by its Accession number, Antigenic cluster, Isolation time, Country and Strain name. This could be achieved by inputting the information of your interest into the textbox below the header of the table (in red box in the figure below). Search results would be returned in a few seconds after selecting the filters by clicking the coin “Y” beside the textbox (indicated by the red arrow).
12. How to cite us?
If you use this server, please cite us in your paper as the following: Youson Peng, Lei Yang, Honglei Li, Yuanqiang Zou, Lizong Deng, Aiping Wu, Xiangjun Du, Dayan Wang, Yuelong Shu, Taijiao Jiang. PREDAC-H3: A user-friendly platform for antigenic surveillance of human influenza A(H3N2) virus based on hemagglutinin sequence. Bioinformatics 2016 32: 2526-2527.
Reference
1 Smith, D. J., et al. (2004). "Mapping the antigenic and genetic evolution of influenza virus." Science 305(5682): 371-376.
2 Du, X., et al. (2012). "Mapping of H3N2 influenza antigenic evolution in China reveals a strategy for vaccine strain recommendation." Nat Commun 3: 709.
3 Bao, Y. M., et al. (2008). "The influenza virus resource at the national center for biotechnology information." J Virol 82(2): 596-601.